Research team "Molecular Neurodegeneration"
Principal Investigator: Prof. Claus U. Pietrzik
Our research team focuses on the molecular and biochemical basis of neurodegeneration in Alzheimer’s disease (AD) serving as a model system. Using cell cultures and transgenic mice models, we analyze the molecular characteristics that cause or affect the progression of AD. We investigate both biochemical processes during the disease’s onset and progression as well as possible new therapeutic approaches.AD is a most common form of dementia, characterized by a gradual cognitive decline and memory loss. Causative for its onset is the loss of synapses and an increased neuronal apoptosis. The pathological appearance consistent with neurodegeneration shows intracellular, neurofibrillary tangles and extracellular senile plaques in the brains of affected individuals.
According to World Health Organization Estimates, 55 million people are affected by dementia. That number is estimated to increase to 65 million by 2030 and 152 million by 2050. Apart from AD, cases of other cerebrovascular diseases such as Parkinson’s disease and multiple sclerosis are steadily increasing due to medical progress and extended life expectancy. To date, curing of the most diseases of the CNS is mostly limited, as the majority of developed drugs are not able to traverse the BBB and enter the brain.
Our team currently focuses on these major topics:
1. The processing of the amyloid precursor protein (APP) by meprin beta and its implications in Alzheimer’s disease.
Senile plaques, the key pathological hallmark of AD, are mainly composed of amyloid beta peptides (Aβ), which are cleavage products of the amyloid precursor protein (APP). The physiological function of APP and its family members APLP1 and APLP2 remain poorly understood. The generation of Aβ peptides represents a complex process involving several proteolytic enzymes called secretases. Two APP secretases: β-secretase and γ-secretase, have been widely investigated, yet therapeutic attempts to block their activity and hence reduce Aβ burden in the brain have been unsuccessful so far [Wong et al., Nature, 2001].
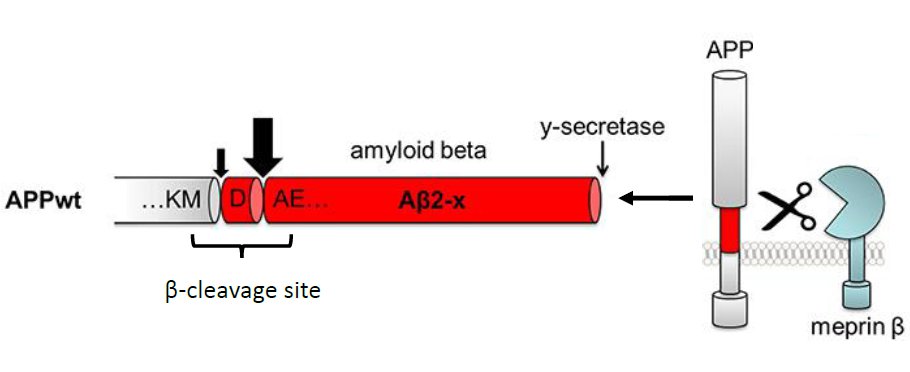
Using proteome analyses and cell culture experiments, we recently confirmed that APP is a substrate for another proteolytic enzyme: meprin β with a cleavage site in close proximity to β-secretase [Fig.1; Becker-Pauly & Pietrzik, Frontiers in Mol. Neuroscience, 2017]. Meprin β has been identified as a potential risk gene associated with AD and meprin β mRNA levels were shown to be increased in brains of AD patients compared to age-matched controls [Patel et al., Neuropathol Appl Neurobiol, 2018; Medoro et al., J Alzheimer’s Dis, 2019]. In one of our recent projects, we are investigating the functional role of meprin β in AD using the transgenic APP London mouse model. APP London (APP/lon) transgenic mice express human APP (isoform 695) with the London mutation (V717I) localized near the γ-secretase cleavage site, therefore not affecting meprin β cleavage site.
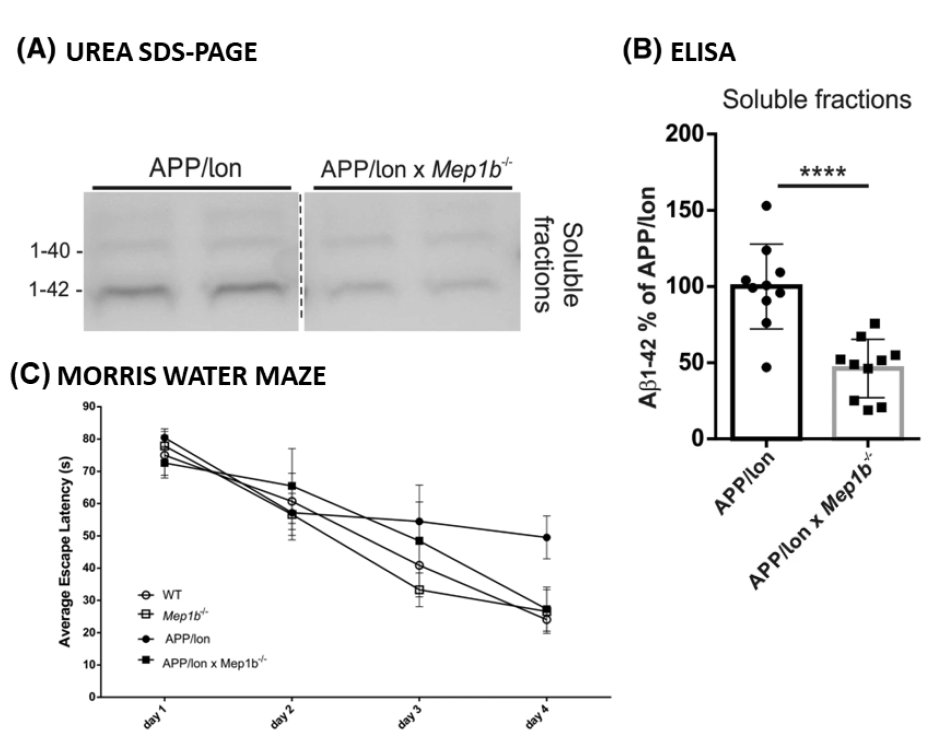
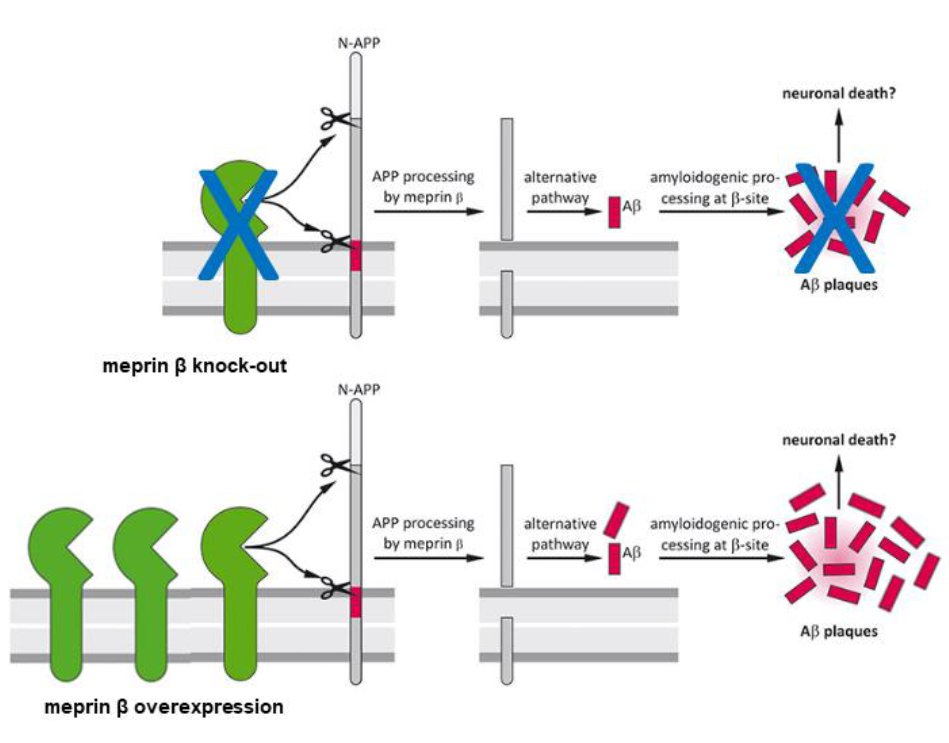
To examine the role of meprin β in AD pathology, we first generated APP/lon mice with homozygous meprin β knock-out. Our studies showed significant reduction of different Aβ species in the brain of these animals compared to controls. Moreover, we observed significant improvement in memory formation in APP/lon mice lacking meprin β expression [Fig. 2; Marengo et al., Cell Mol. Life Sci., 2022]. To further investigate the role of meprin β in AD, we established the APP /lon mouse model with a neuron-specific overexpression of meprin β. With this approach we aim not only to investigate whether meprin β overexpression affects premature-onset of pathology, but also to identify possible additional brain derived substrates of meprin β. To fully understand different aspects of meprin β role in AD we are cooperating with several research groups to be able to create a detailed picture of the role of meprin β in the brain (Fig. 3).
The blood-brain barrier (BBB) plays a crucial role in maintaining the brain homeostasis, preventing the brain from uncontrolled entrance or diffusion of potentially harmful molecules. Several recent studies have also highlighted the importance of the BBB in neurodegenerative diseases such as Alzheimer's disease (AD) by regulating transport of the amyloid beta (Aβ) peptide via the low-density lipoprotein receptor related protein 1 (LRP1). Therefore, we are interested in the role of LRP1 as key receptor in Aβ transport across the blood-brain-barrier (BBB).
We have established a transgenic mouse model strain with inducible brain endothelial -specific deletion of LRP1 (Lrp1BE-/-) and crossed it with 5xFAD mouse model of AD (5xFAD Lrp1BE-/-) to evaluate LRP1-mediated Aβ peptide clearance across the BBB in vivo. In our recent findings [Fig.4; Storck et al., J Cli Inv, 2016], we demonstrated that LRP1 deletion in 5xFAD mice resulted in reduced plasma Aβ levels and elevated soluble brain Aβ, aggravating the cognitive decline. Furthermore, using LRP1 knock-out mice, we investigated the precise mechanism by which LRP1 transports the Aβ across the BBB. We showed that LRP1 expression positively correlates with ABCB1/Pgp efflux transporter at the BBB [Storck et al., Brain Behav Immun, 2018]. Moreover, we found out that Aβ is removed from the brain by cooperation between two BBB transporters: LRP1 and ABCB1/P-gp, mediated by phosphatidylinositol binding clathrin assembly protein (PICALM).
In our recent findings, we investigated a role of LRP1 in maintaining the BBB integrity [Storck, Kurtyka & Pietrzik, Fluids Barriers CNS, 2021]. Using generated by us LRP1 knock-out mice (Lrp1BE-/-), we demonstrated metalloprotease-mediated tight junctions’ degradation, increased BBB permeability and shrinkage of the brain manifested by reduced water content (Fig. 5).
We have established a transgenic mouse model strain with inducible brain endothelial -specific deletion of LRP1 (Lrp1BE-/-) and crossed it with 5xFAD mouse model of AD (5xFAD Lrp1BE-/-) to evaluate LRP1-mediated Aβ peptide clearance across the BBB in vivo. In our recent findings [Fig.4; Storck et al., J Cli Inv, 2016], we demonstrated that LRP1 deletion in 5xFAD mice resulted in reduced plasma Aβ levels and elevated soluble brain Aβ, aggravating the cognitive decline. Furthermore, using LRP1 knock-out mice, we investigated the precise mechanism by which LRP1 transports the Aβ across the BBB. We showed that LRP1 expression positively correlates with ABCB1/Pgp efflux transporter at the BBB [Storck et al., Brain Behav Immun, 2018]. Moreover, we found out that Aβ is removed from the brain by cooperation between two BBB transporters: LRP1 and ABCB1/P-gp, mediated by phosphatidylinositol binding clathrin assembly protein (PICALM).
In our recent findings, we investigated a role of LRP1 in maintaining the BBB integrity [Storck, Kurtyka & Pietrzik, Fluids Barriers CNS, 2021]. Using generated by us LRP1 knock-out mice (Lrp1BE-/-), we demonstrated metalloprotease-mediated tight junctions’ degradation, increased BBB permeability and shrinkage of the brain manifested by reduced water content (Fig. 5).


Our current research aims to investigate the functional role of LRP1 as a transporter of therapeutics to the brain. LRP1 expression in the brain decreases during ageing and even more prominent decline is observed in AD. Therefore, for successful LRP1-mediated delivery of drugs, LRP1 expression must be increased. Using brain endothelium-specific adeno-associated virus [Körbelin et al, EMBO, 2016], we attempt to overexpress truncated LRP1mini receptors at the BBB and boost clearance of Aβ peptides from the brain as well as deliver therapeutics to the CNS (Fig. 6).

To analyze the lipoprotein receptor transport across the BBB, we are investigating post-transcriptional regulatory mechanisms of proprotein convertase subtilisin/kexin type 9 (PCSK9), a member of the proteinase K subfamily of subtilases. In our previous work, we identified circulating PCSK9 as a regulator of LRP1 mediated Aβ transport across the BBB [Fig.7; Mazura et al., Cell Mol Life Sci, 2022]. By using an established BBB in vitro model, we observed significantly reduced LRP1 mediated Aβ transport across an endothelial monolayer after treatment with recombinant PCSK9. Consistently, we reversed this outcome in vivo by repetitive peripheral administration of FDA-approved monoclonal anti-PCSK9 antibodies in the 5xFAD mouse model. Anti-PCSK9 antibody intraperitoneal treatment resulted in substantially decreased Aβ concentrations in the CNS (Fig. 8) and improved hippocampus-dependent learning behavior compared to control treated mice (Fig. 9). Since peripheral PCSK9 inhibition was insufficient to reproduce these effects in 5xFAD Lrp1 BE-/- mice, our data might reveal the potential to modify BBB receptor quantities in therapeutic contexts.

A: LRP1 deletion at the endothelium of the BBB results in increased Aβ levels in the brain [Storck et al., J Cli Inv, 2016]. B: PCSK9 regulates LRP1 levels under physiological conditions through binding at the blood site, which leads to the lysosomal degradation of LRP1 (B left). PCSK9 inhibitor treatment results in increased LRP1 levels at the BBB and subsequent increased Aβ clearance across the BBB (B right) [Mazura et al., Cell Mol Life Sci, 2022].

Soluble (A + B) and insoluble (C + D) brain Aβ1–40 (A + C) or Aβ1–42 (B + D) level of 6-month-old 5xFAD or 5xFAD LRP1BE-/- mice, treated, repetitively, with 1 µg/g anti-PCSK9 antibody or 0.9% NaCl for 10 weeks were assessed via ELISA.
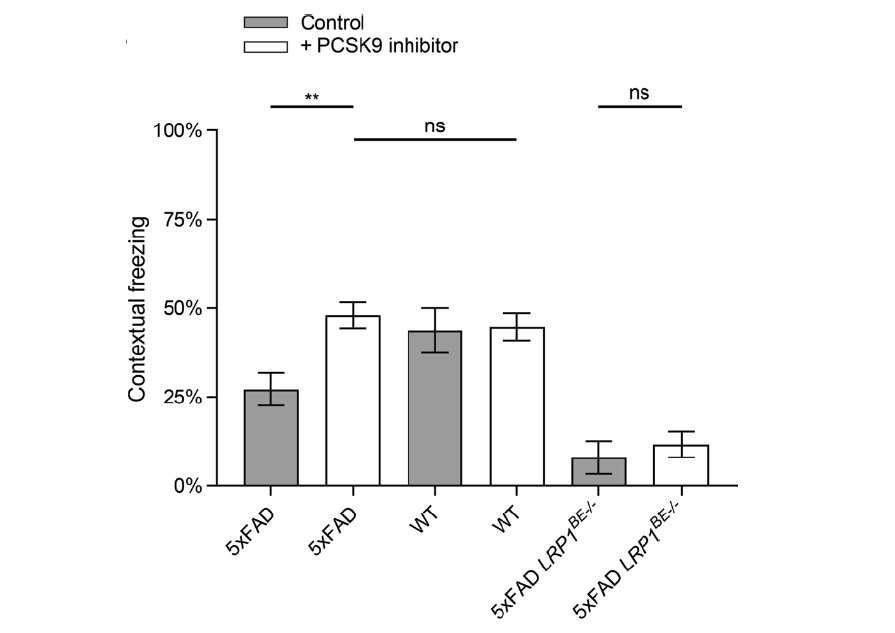
transgenic mice in a Fear Conditioning paradigm. 6-month-old 5xFAD mice treated with 1 µg/g anti PCSK9 antibody or 0.9% NaCl for 10 weeks were analyzed for hippocampus-dependent contextual fear response in comparison to wildtype and LRP1BE-/- littermates. Fear was assessed by freezing behavior (the absence of all but respiratory movements), when confronted again to the context of the training session and displayed as percentage of the measurement period. Higher freezing behavior percentage indicates stronger memory formation.
Besides the pharmacological approach to reduce PCSK9 functionality in vivo, we want to investigate the impact of a complete absence of PCSK9 on BBB function and Aβ clearance. Therefore, we crossed PCSK9 knock-out mice into the 5xFAD model of AD with tamoxifen-inducible endothelial -specific deletion of LRP1 (5xFAD Lrp1BE-/- Pcsk9-/-). We will determine the expression of LRP1 and its role on clearance of Aβ-peptides from the brains of AD mice. Additionally, we aim at inducing a brain endothelial specific knock-out of LRP1 in the 5xFAD Lrp1BE-/- Pcsk9-/- mouse model to prove that PCSK9-regulated Aβ clearance depends on brain endothelial LRP1. The genetic alteration of PCSK9 expression in an AD mouse model will furthermore help us to explain the mechanism behind the beneficial effect of PCSK9 inhibitors, which we have reported before [Mazura et al., Cell Mol Life Sci, 2022].
Selected publications:
Pflanzner T et al. 2010 Curr Alzheimer Res.
Pflanzner T et al. 2011 Neurobiol Aging
Pflanzner T et al. 2012 J Cereb Blood Flow Metab.
Wagner S et al. 2012 PLoS One.
Maier W et al. 2013 Mol Neurodegener.
Meister S et al. 2013 Alzheimer's Research & Therapy
Martínez Vera NP et al. 2014 PLoS One.
Meister S et al. 2015 J Cereb Blood Flow Metab.
Storck SE et al. 2016 J Clin Invest.
Afrimzon E et al. 2016 Journal of Nanoparticle Res
Eggert S et al. 2017 Cell Mol Life Sci.
Zandl-Lang M et al. 2017 Biochim Biophys Acta.
Storck SE, Pietrzik CU. 2017 Pharm Res.
Di Spiezio A et al. 2017 Mol Metab.
Storck SE et al. 2018 Brain Behav Immun.
Van Gool B et al. 2019 Mol Neurobiol.
Marques ARA et al. 2019 Autophagy
Gindorf M et al. 2021 J Cereb Blood Flow Metab.
Sandin ES et al. 2021 Int J Mol Sci.
Storck SE et al. 2021 Fluids Barriers CNS
Storck SE et al. 2022 Handb Exp Pharmacol.
Mazura AD et al. 2022 Cell Mol Life Sci.
Selected publications:
Pflanzner T et al. 2010 Curr Alzheimer Res.
Pflanzner T et al. 2011 Neurobiol Aging
Pflanzner T et al. 2012 J Cereb Blood Flow Metab.
Wagner S et al. 2012 PLoS One.
Maier W et al. 2013 Mol Neurodegener.
Meister S et al. 2013 Alzheimer's Research & Therapy
Martínez Vera NP et al. 2014 PLoS One.
Meister S et al. 2015 J Cereb Blood Flow Metab.
Storck SE et al. 2016 J Clin Invest.
Afrimzon E et al. 2016 Journal of Nanoparticle Res
Eggert S et al. 2017 Cell Mol Life Sci.
Zandl-Lang M et al. 2017 Biochim Biophys Acta.
Storck SE, Pietrzik CU. 2017 Pharm Res.
Di Spiezio A et al. 2017 Mol Metab.
Storck SE et al. 2018 Brain Behav Immun.
Van Gool B et al. 2019 Mol Neurobiol.
Marques ARA et al. 2019 Autophagy
Gindorf M et al. 2021 J Cereb Blood Flow Metab.
Sandin ES et al. 2021 Int J Mol Sci.
Storck SE et al. 2021 Fluids Barriers CNS
Storck SE et al. 2022 Handb Exp Pharmacol.
Mazura AD et al. 2022 Cell Mol Life Sci.