Research Interests
The Kindler lab is part of the Department of Hematology, Oncology, and Pneumology at the University Medical Center of the Johannes Gutenberg-University of Mainz, Germany.
In our laboratory, we are interested in understanding signal transduction pathways in cancer cells, with a focus on hematopoietic malignancies. A detailed understanding of aberrant signaling should provide insights into the process of malignant transformation, mechanisms of drug resistance and vulnerability of cancer cells. In addition, the identification of functional differences between cancer stem cells and their normal counterparts will allow us to develop novel therapeutic strategies. In this context we use a variety of distinct experimental approaches and tumor models, including cell culture, defined mouse models (e.g. Flt3-ITD knock-in, conditional KrasG12D knock-in, bone marrow transplantation) and primary tumor samples.
Our laboratory is located in a scientifically stimulating environment and broadly integrated in the research community of the Johannes Gutenberg-University.
If you are interested in an application for a thesis (medical or master) please do not hesitate to contact us. Students with a strong interest in hematology/oncology are always welcome to join the lab for rotations.
Current Projects
Based on our current understanding, it is believed that the leukemic clones are perpetuated by a rare population of leukemia stem cells (LSC). These LSCs share many characteristics with normal hematopoietic stem cells (HSCs), such as phenotype, self-renewal potential, a quiescent cell cycle status and enhanced drug resistance. Whereas cytotoxic drugs eradicate actively cycling leukemic blasts, they often do not target LSCs embedded in the bone marrow niche. Recent studies indicate that the bone marrow stromal cells and the hematopoietic microenvironment in the bone marrow dictate stem cell quiescence, lack of motility and expression of anti-apoptotic proteins which leads to LSC survival and ultimately results in refractory leukemic disease or relapse. The disruption of specific LSC–stroma cell interactions might drive this leukemia-maintaining cell population into cell cycle and thus render them vulnerable to chemotherapy or targeted therapies.
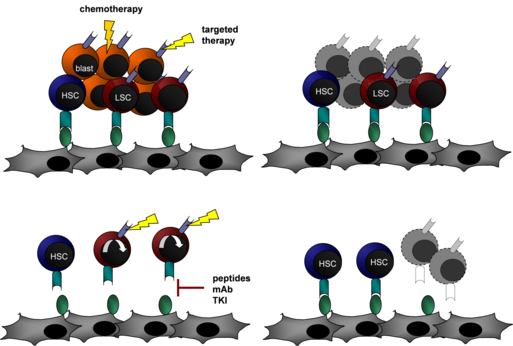
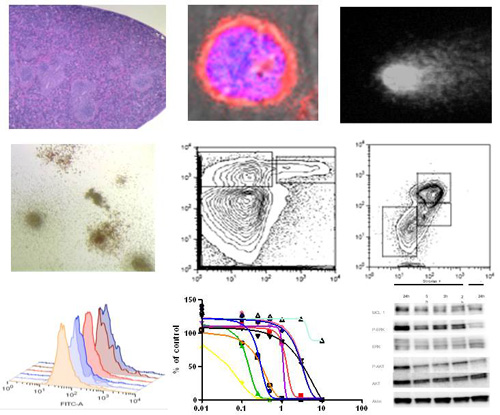
In order to identify genes regulated upon leukemia–stroma cell interaction we performed global gene expression screens in stromal feeder layer cells and leukemic blasts co-cultured for different time periods. In comparison to mono-cultured blasts or normal HSCs, several differentially regulated genes such as receptors, ligands, extracellular matrix components or known proto-oncogenes were identified in the gene expression analysis of co-cultured leukemic blasts. We currently explore the role of several candidate genes and the associated signaling pathways. Promising target genes are further evaluated in vivo in murine leukemia models. In addition we are performing a comparative functional screen using a genome-scale CRISPR-Cas9 knockout library in leukemic blasts mono-cultured or co-cultured with stromal cells aiming to identify genes involved in stroma-mediated drug resistance.
Using a conditional KrasG12D knock-in mouse model, we and others have demonstrated that expression of oncogenic Kras in hematopoietic progenitor cells causes an arrest at the DN2/3 stage during T-cell differentiation followed by the development of an aggressive T-cell lymphoblastic leukemia/lymphoma with long disease latency. Interestingly, 50% of analysed leukemia samples harboured Notch1-mutations, likely acquired during the KrasG12D-mediated block in differentiation. These additional mutations may be acquired by replicative stress or increased reactive oxygen species (ROS) production. Alternatively, oncogenic RAS may directly affect DNA repair pathways, thereby causing misrepair and, due to concomitant resistance to apoptotic cell death, accumulation of genomic changes. In line with this hypothesis, recent studies indicated that DNA damage response and repair (DDR) can be disturbed within a defined malignant background. Using several in vitro and in vivo models, we analyse DNA repair pathways in KRASMUT and KRASWT cells. Upon induction of DNA double strand breaks, expression of oncogenic KRAS was associated with a shift to the preferential use of the error-prone alternative non-homologous end-joining (alt-NHEJ) pathway.
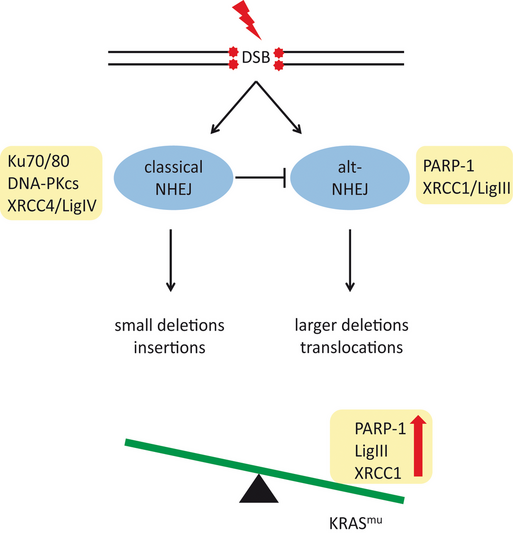
Interestingly, targeting components of this pathway significantly enhanced chemotherapy-induced apoptotic cell death. Of note, this effect was specific for leukemic cell harbouring KRAS-mutations. Currently, we systematically explore DNA damage repair pathways in the context of distinct oncogenes in several leukemia models and solid tumors.
Acute myeloid leukemia (AML) is characterized by aberrant signal transduction resulting in enhanced cell survival, proliferation and resistance to apoptotic cell death. Rewired signaling is caused by acquired genetic alterations and/or by overexpression/aberrant expression of signal transduction molecules. Whereas canonical signal transduction initiates transcription factor activation and target gene expression, recent reports have also shown profound effects on chromatin modification and regulation of DNA damage response and repair. Of note, disturbed signal transduction in cancer cells results in tumor specific alterations associated with aberrant self-renewal and stress response but also synthetic vulnerability. Against this background we aim to dissect the influence of defined alterations within signal transduction pathways on epigenetic modifications, DNA repair and resistance to genotoxic agents/small molecule inhibitors. To address this question, we take advantage of a broad spectrum of mouse models. In addition, we have created several isogenic cell culture models using siRNA mediated gene knockdown or CRISPR-Cas9-mediated knockout. Further, cancer cells will be treated with specific small molecule inhibitors targeting signal transduction proteins. The impact on DNA damage response and repair will be explored by applying qRT-based gene expression analyses and in vivo DNA repair assays. Based on these technologies we hope to identify novel treatment strategies combining inhibitors that target DNA damage repair pathways, epigenetic modifiers and signal transduction molecules.
Acute myeloid leukemia (AML) is characterized by increased proliferation, evasion of apoptotic stimuli and block of differentiation. Mutations in FMS-like tyrosine kinase 3 (FTL3) receptor such as the internal tandem duplicate (FLT3-ITD) are found in about 25% of de novo AML cases. This mutation causes constitutive active downstream signaling and allows cells to replicate at a high proliferation rate. The question we like to address is how cells maintain genomic integrity in such conditions.
Poly (ADP-ribose) polymerase (PARP) is a family of proteins that are involved in various genome maintenance processes and play a major role in correcting single strand breaks (SSB). Perturbing highly proliferating cells by inhibiting PARP can induce genetic instability by increasing the number of double strand breaks (DSB). The recruitment of DSB repair pathway proteins to the site of damage can be quantified by an accumulation of γH2AX foci.
In this project we aim at shedding light in mechanisms involved in maintaining genomic stability, exploring pathways regulating DSB repair and exploit mechanisms that prevent induction of apoptosis in highly proliferating AML cells.
Publications (Selection)

For a complete list of publications please click on the ResearcherID button. There also citation metrics can be found.
- Sasca D, Hähnel PS, Szybinski J, Khawaja K, Kriege O, Pante SV, Bullinger L, Strand S, Strand D, Theobald M, Kindler T. SIRT1 prevents genotoxic stress-induced p53 activation in acute myeloid leukemia. Blood. 2014;124(1):121-33.
- Hähnel PS, Enders B, Sasca D, Roos WP, Kaina B, Bullinger L, Theobald M, Kindler T. Targeting components of the alternative NHEJ pathway sensitizes KRAS mutant leukemic cells to chemotherapy. Blood. 2014;123(15):2355-66.
- Hartwell KA, Miller PG, Mukherjee S, Kahn AR, Stewart AL, Logan DJ, Negri JM, Duvet M, Järås M, Puram R, Dancik V, Al-Shahrour F, Kindler T, Tothova Z, Chattopadhyay S, Hasaka T, Narayan R, Dai M, Huang C, Shterental S, Chu LP, Haydu JE, Shieh JH, Steensma DP, Munoz B, Bittker JA, Shamji AF, Clemons PA, Tolliday NJ, Carpenter AE, Gilliland DG, Stern AM, Moore MA, Scadden DT, Schreiber SL, Ebert BL, Golub TR. Niche-based screening identifies small-molecule inhibitors of leukemia stem cells. Nat. Chem. Biol. 2013;9(12):840-8.
- Cabezas-Wallscheid N, Eichwald V, de Graaf J, Löwer M, Lehr HA, Kreft A, Eshkind L, Hildebrandt A, Abassi Y, Heck R, Dehof AK, Ohngemach S, Sprengel R, Wörtge S, Schmitt S, Lotz J, Meyer C, Kindler T, Zhang DE, Kaina B, Castle JC, Trumpp A, Sahin U, Bockamp E. Instruction of haematopoietic lineage choices, evolution of transcriptional landscapes and cancer stem cell hierarchies derived from an AML1-ETO mouse model. EMBO Mol. Med. 2013;5(12):1804-20.
- Tam WF, Hähnel P, Schüler A, Lee BH, Okabe R, Zhu N, Pante S, Raffel G, Mercher T, Wernig G, Bockamp E, Kreft A, Robinson GW, Hennighausen L, Gilliland DG, Kindler T. STAT5 is crucial for the maintenance of leukemic stem cells in MOZ-TIF2 induced acute myeloid leukemogenesis. Cancer Research. 2013;73(1):373-84.
- Breitenbuecher F, Markova B, Kasper S, Carius B, Stauder T, Schnittger S, Haferlach T, Boehmer F, Huber C, Kindler T*, Fischer T*. Rewired FLT3_ITD Signaling Pathways Confer Primary Resistance to FLT3-Kinase Inhibitors in Acute Myeloid Leukemia. Blood. 2009;23;113(17):4063-73. * Equal contribution
- Kindler T, Cornejo MG, Scholl C, Liu J, Leeman DS, Haydu JE, Fröhling S, Lee BH, Gilliland DG. K-RasG12D-induced T-cell lymphoblastic lymphoma/leukemias harbor Notch1 mutations and are sensitive to γ-secretase inhibitors. Blood. 2008;112(8):3373-82.
- Mercher T, Cornejo MG, Sears C, Kindler T, Moore SA, Maillard I, Pear WS, Aster JC, Gilliland DG. Notch Signaling Specifies Megakaryocyte Development from Hematopoietic Stem Cells. Cell Stem Cell. 2008;3(3):314-26.
- Kindler T, Breitenbuecher F, Kasper S, Estey E, Giles F, Feldman E, Ehninger G, Schiller G, Klimek V, Nimer SD, Gratwohl A, Choudhary CR, Mueller-Tidow C, Serve H, Gschaidmeier H, Cohen PS, Huber C, Fischer T. Identification of a novel activating mutation (Y842C) within the activation loop of FLT3 in patients with acute myeloid leukemia (AML). Blood. 2005;105(1):335-40.
- Kindler T, Breitenbuecher F, Kasper S, Stevens T, Carius B, Gschaidmeier H, Huber C, Fischer T. In BCR-ABL-positive cells, STAT-5 tyrosine-phosphorylation integrates signals induced by imatinib mesylate and Ara-C. Leukemia. 2003;17(6):999-1009.
Kindler Laboratory's Team
Principal Investigator
Univ.-Prof. Dr. Thomas Kindler
Tel.: 06131 17-5046
E-Mail: Thomas.Kindler@unimedizin-mainz.de
Laboratory Members
Alumni
- Sven Henninger, M. Sc.
- Dr. med. Undine Lange
- Dr. rer. nat. Saskia Pante
- Dr. med. Daniel Sasca
- Dr. rer. nat. Andrea Schüler
- Dr. rer. nat. Torsten Stauder, M. Sc.
- Jakub Szybinski Guerrero
