Für welche Erkrankungen ist eine PID möglich?
Monogene Erkrankung
Unter einer monogenen Erkrankung versteht man eine den Mendel‘schen Erbregeln folgende familiäre Erkrankung, deren Ursache in der Veränderung eines einzelnen Gens liegt. Nach heutigem Kenntnisstand sind schätzungsweise etwa 8000 verschiedene Erkrankungen monogen erblich bedingt. Die Erkrankung kann entweder autosomal dominant, autosomal rezessiv oder X-chromosomal vererbt werden.Praktische Durchführung einer PID (PGT-M) bei monogenen Erkrankungen
Grundsätzlich besteht durch eine PID (PGT-M) die Möglichkeit, Embryonen eines Paares mit hohem Risiko für das Auftreten einer schwerwiegenden Erbkrankheit auf die familiäre genetische Veränderung hin zu untersuchen. Dazu erfolgt die Analyse einer Trophektodermbiopsie des Embryos mithilfe verschiedener, der Fragestellung angepasster molekulargenetischer Labormethoden. Dieser Untersuchung gehen aufwändige genetische Voruntersuchungen zur Etablierung und Validierung dieses Testsystems voraus. Unsere Untersuchungen beinhalten ein diverses Methodenportfolio wie Mikrosatellitenanalysen, Fragmentanalysen sowie Sanger- und Pyrosequenzierung und werden in unserem nach Norm DIN EN ISO 15189:2014 akkreditierten Labor durchgeführt. Die jeweilige Methodik wird basierend auf der individuellen spezifischen Fragestellung und der vorliegenden Form der zu untersuchenden Genveränderung ausgewählt. Dazu sind Blut- oder DNA-Proben des Paares sowie gegebenenfalls Proben weiterer Familienangehöriger oder eines betroffenen Kindes notwendig. Für die Beurteilung der Entwicklungsfähigkeit nicht betroffener Embryonen kann eine zusätzliche Beurteilung von Aneuploidien (PGT-A) bei der Ethikkommission beantragt werden. Damit eine PID durchgeführt werden kann, muss die ursächliche genetische Veränderung für eine in der Familie aufgetretene Erkrankung bekannt sein.Strukturelle Chromosomenveränderungen
Wir Menschen tragen von all unseren Chromosomen 2 Ausfertigungen – eine wird von der Mutter und die andere vom Vater ererbt. Im Rahmen der Keimzellentwicklung (Entstehung von Samen- und Eizellen) ist es ein normaler Prozess, dass zueinander passende (homologe) Chromosomen Stücke austauschen, um die genetische Vielfalt bei den Nachkommen zu erhöhen. Dabei kann es passieren, dass bei diesem Austausch chromosomale Bruchteile zwischen zwei nicht zueinander passenden (nicht homologen) Chromosomen ausgetauscht werden, was wir als balancierte Translokation bezeichnen. Dabei werden zwei Formen unterschieden:Reziproke Translokation (Häufigkeit etwa 1:700 Neugeborene):
Reziproke Translokationen entstehen durch je ein Bruchereignis in 2 Chromosomen mit gegenseitigem Austausch der Fragmente (siehe Abbildung 1: Darstellung 2). In den meisten Fällen führen diese Umlagerungen nicht zu einem Verlust von chromosomalem Material, sodass die überwiegende Mehrheit der Träger klinisch unauffällig ist. Die Keimzellbildung (Eizellen oder Spermien) ist bei reziproken Translokationen störungsanfällig, weil sich im Bereich der Translokation die Chromosomen nicht ohne weiteres paaren können. Im Rahmen der Zellteilung ordnen sich die zueinander passenden Chromosomen an der Teilungsebene der Zelle an, indem sich die einander entsprechenden (homologen) Abschnitte paarweise zusammenfinden. In den Abschnitten, die von der Translokation betroffen sind, liegen keine Homologien der Chromosomen vor. Deshalb sind die involvierten Chromosomen gezwungen, sich in anderer Form aneinander zu lagern. Im Zuge der Trennung der Chromosomen bei der Keimzellreifung besteht daher das Risiko, dass die Chromosomen so verteilt sind, dass Abschnitte zu viel oder zu wenig vorhanden sind (unbalancierte Translokation), wodurch ein Risiko für Kinder mit teilweise schweren Gesundheitsstörungen besteht.
Robertson'sche Translokation (Häufigkeit etwa 1:1.000 Neugeborene):
Eine Robertson’sche Translokation (Abbildung 1: Darstellung 4) kann nur zwischen bestimmten Chromosomen auftreten - den sogenannten akrozentrischen Chromosomen (13, 14, 15, 21 und 22). Wir unterteilen Chromosomen in einen kurzen (p-) und einen langen (q-) Arm, die durch das sogenannte Zentromer voneinander getrennt sind. Bei akrozentrischen Chromosomen ist der kurze Arm sehr klein und enthält nur wenig Gene, die zudem in vielfacher Kopie im Genom vorliegen. Im Rahmen der Keimzellentwicklung kann es passieren, dass sich die beiden langen Arme der betroffenen Chromosomen verbinden. Auch die beiden kurzen Arme verbinden sich zu einem Chromosom, welches jedoch in der Regel verloren geht. Dieser Verlust hat aber nach heutigem Kenntnisstand keine Auswirkungen auf den klinischen Phänotyp des Betroffenen.. Etwa 75% aller Robertson'schen Translokationen betreffen je ein Chromosom 13 und ein Chromosom14 rob(13;14). Diese Veränderung findet sich häufiger bei Paaren mit unerfülltem Kinderwunsch. Bei Männern ist die Translokation mit verminderten Spermienzahlen (Oligozoospermie) assoziiert. Bei weiblichen Translokationsträgern ist das Risiko für eine Fehlgeburt (Wahrscheinlichkeit 28%) und eine Schwangerschaft mit einer Trisomie 13 (Pätau-Syndrom, Wahrscheinlichkeit unter 10%) erhöht.
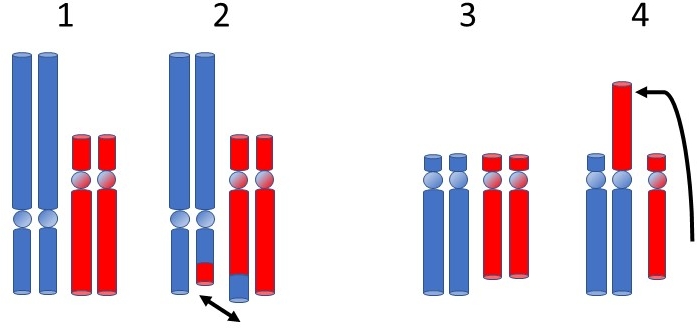
Abbildung 1: strukturelle Chromosomenveränderungen: 1) unverändertes Chromosomenpaar 2) reziproke Translokation 3) unverändertes akrozentrisches Chromosomenpaar 4) Robertson'sche Translokation
Praktische Durchführung einer PID (PGT-SR) bei strukturellen Chromosomenveränderungen
Zum Ausschluss möglicher unbalancierter Chromosomenveränderungen im Embryo als Folge einer reziproken oder Robertson'schen Translokation bei einem Elternteil verwenden wir eine Next-Generation-Sequencing-basierte genomweite Imbalancekartierung. Gleichzeitig kann mit dieser Methode die Anzahl aller anderen Chromosomen im Hinblick auf mögliche Fehlverteilungen (Aneuploidien; PGT-A) beurteilt werden.
Abbildung 2: Auffällige strukturelle Chromosomenveränderung der Trophektodermbiopsie eines Embryos

Abbildung 3: Unauffälliger Chromosomenstatus der Trophektodermbiopsie eines Embryos